Delimiting a region of interest
Jacob Marsh
2023-03-08
Source:vignettes/articles/Delimiting_region.Rmd
Delimiting_region.RmdIntroduction
Local haplotyping first and foremost requires the specification of a genomic region to analyse. The fundamental aim of delimiting a genomic region to haplotype with crosshap should be to include the minimum range of sites that sufficiently captures groups of linked sites that are relevant to a trait or feature of interest.
There is no universally ‘correct’ way to delimit your region of interest, it should instead be decided upon after carefully considering your dataset, the purpose of your analysis, and the genomic features surrounding a prospective site. Here, we’ll have a look at a few possible methods that could be used to delimit a region of interest.
Method 1: Range of significant SNPs from GWAS
Trait-associated regions identified through GWAS are particularly suitable for analysis with crosshap as the phenotypic data used as input can be directly used as input for crosshap to aid in capturing and characterizing informative haplotypes. However, crosshap characterises a specific region of interest, where GWAS only provides individual trait associated loci.
General (GLM) and mixed linear (MLM) GWAS models provide association results for all genomic sites, which allows the user to delimit a genomic region by including all variants between the most distal loci possessing a minimum level of significance. If your GWAS method combines linked SNPs into single loci (e.g. FarmCPU), then you can disentangle this by calculating pairwise linkage between all SNPs in a wider region and delimit it based on the furthest loci above a given threshold (e.g. R^2 with GWAS SNP > 0.8).

This method will result in a region that contains all highly significant SNPs that are highly associated with a given trait.
Method 2: Haplotype block boundaries
Sometimes you may be more interested in analysing the linkage
structures between variants agnostic of trait association information.
In these cases a visualized LD matrix (e.g. using LDBlockShow) or the
boundaries of a haplotype block (e.g. defined using
Haploview)
may be useful for delimiting a genomic region of interest.
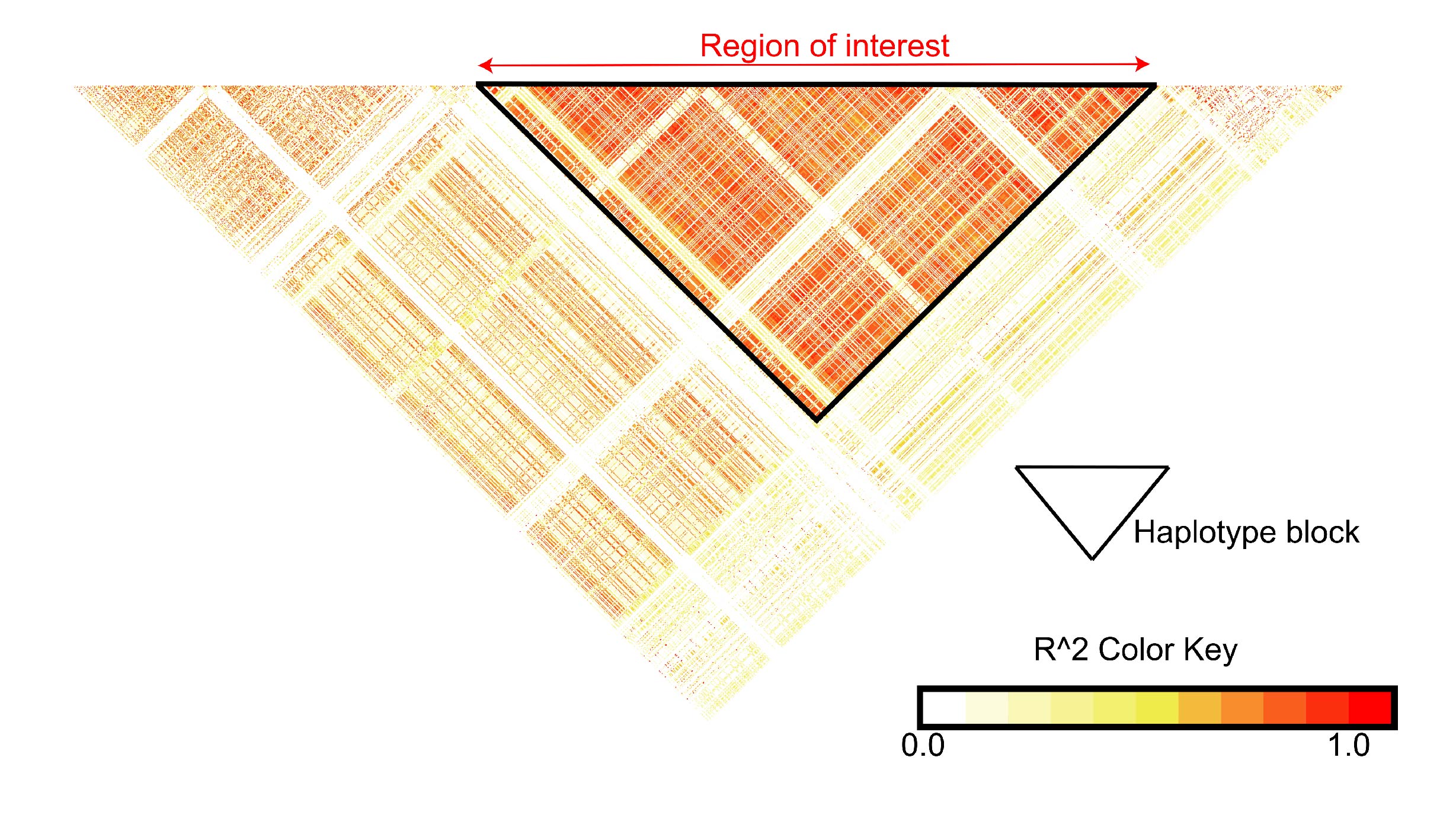
This method will result in a region with high homogeneous internal linkage relative to distal sites and will ensure that you’re haplotyping a region with strong linkage patterns.
Method 3: Gene cluster
The co-location of two or more gene structures or independent variants of interest can be useful for delimiting a region to explore linkage patterns that span them. If there is a gene cluster you’re interested in characterising, the positions of these features can directly be used as the boundaries for your region of interest. It can be useful to use this method alongside testing of general linkage in the region (Method 2).

This method will result in a region that contains relevant genomic features, however this may not include with the breadth of linkage patterns (i.e. Marker Groups may extend beyond the region analysed).
What now?
After figuring out the boundaries of your region, the next step is to subset your VCF to only include SNP information for that region.
To extract the region of interest from your VCF, you can use something like tabix on the command line, just make sure you keep the final header line that starts with ‘#CHR’:
$ grep -m 1 '#CHR' in.vcf > in.region.vcf
$ bgzip in.vcf
$ tabix in.vcf.gz Chr1:100000-200000 >> in.region.vcf
Now your VCF is ready for analysis with crosshap! For more information have a look at the Getting started vignette.