Introduction
Between haplotypes, Marker Groups, allelic combinations and unplaced loci, there’s a lot of data transformation that crosshap performs to the point where it can be difficult to take a step back and appreciate what genomic patterns are shared between individuals with the same assigned haplotype. Here, we’ll make an animated GIF that will help us visualize this information. The GIF doesn’t necessarily reveal more details about our haplotypes, however it can be useful as a sanity check, and to get a better understanding of what our results reflect.
UMAP
Uniform Manifold Approximation Projection (UMAP) is a useful dimension reduction tool for visualizing clustering results. In brief, it will attempt to co-locate points (SNPs) that are correlated (in linkage) in two-dimensional space, similar to PCA.
You’ll need to set several UMAP parameters. The data input for UMAP
can thankfully be taken as the LD matrix provided to crosshap for
haplotyping. n_neighbors should match the MGmin parameter
provided to crosshap for haplotyping. The final two parameters,
min_dist and spread will need some testing to
optimize.
Capturing Marker Groups with UMAP
First lets visualize where our Marker Groups are in the UMAP which can be a useful reference for later, and can help us optimize our UMAP before we use it to map our individuals.
#Run UMAP on LD matrix to get x & y coordinates for plotting
umap_in <- umap::umap(LD, min_dist = 2, spread = 2.5, n_neighbors = 30)
#Add UMAP coordinates to SNPs with Marker Group information
MGumap_data <- umap_in$layout %>% tibble::as_tibble() %>%
cbind(rownames(umap_in$data)) %>% tibble::as_tibble() %>%
dplyr::rename(UMAP1 = V1, UMAP2 = V2, ID = 'rownames(umap_in$data)') %>%
#HapObject$Haplotypes_MGmin30_E0.6$Varfile contains variant information at epsilon = 0.6
dplyr::left_join(HapObject$Haplotypes_MGmin30_E0.6$Varfile %>%
dplyr::select(ID, MGs), by = "ID") %>%
dplyr::mutate(MGs = gsub('0', NA, MGs))
#Plot SNPs by UMAP coordinates, coloured by Marker Group
MGumap <- ggplot2::ggplot(MGumap_data, ggplot2::aes(x = UMAP1, y = UMAP2)) +
ggplot2::geom_point(alpha = 0.4, ggplot2::aes(colour = MGs), size = 1, na.rm = F)+
ggplot2::theme_minimal() +
ggplot2::guides(colour = ggplot2::guide_legend(override.aes = list(size = 5, alpha = 0.7), title = "Marker group"))
MGumap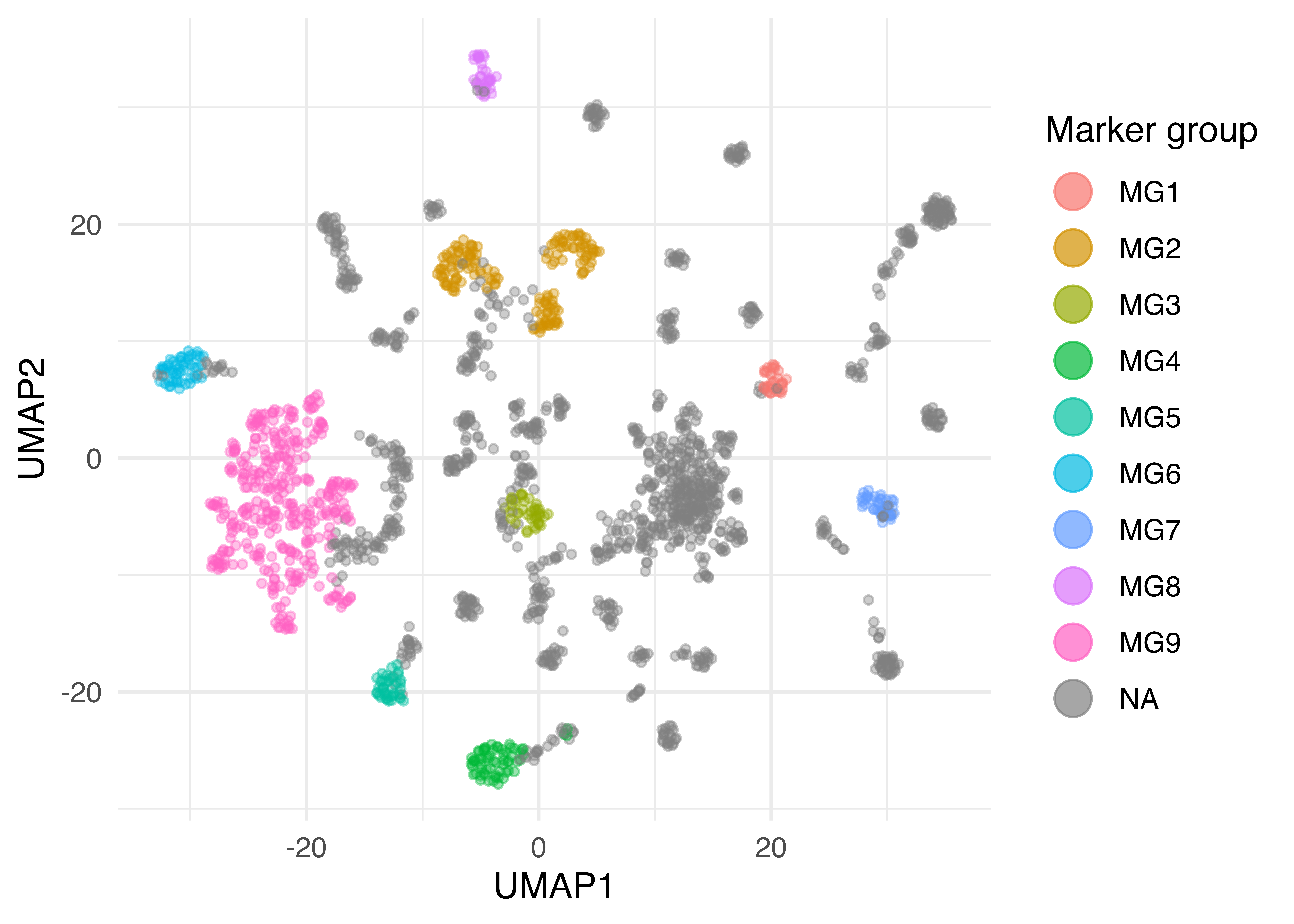
What haplotypes do these Marker Groups define?
crosshap::build_mid_dotplot(HapObject, epsilon = 0.6, hide_labels = F)
A full guide to optimizing UMAP parameters is beyond the scope of
this tutorial, but the MGumap plot we’ve created should be able to help
with that if needed. The goal when optimizing your UMAP is to find
parameters that co-locate SNPs of the same Marker Group in distinct
clusters. Read here
for more information on how to optimize min_dist and don’t
forget to keep spread above min_dist()!
Building UMAP GIF
The prepare_hap_umap() function in crosshap takes UMAP
results and a haplotype object built by run_haplotyping()
with an epsilon resolution and builds a plot for individuals from each
haplotype with their SNP alleles across the UMAP.
What’s the nsamples parameter?
The nsamples parameter will match the number of unique
frames in your final GIF. It can be changed depending on the number of
randomly sampled individuals you’d like to have from each haplotype. 25
is the default, however if you have more individuals it may be worth
increasing, or if it is taking too long to run consider lowering this
value. NOTE: nsamples can include the same individual multiple times
which is useful when nsamples > nIndividuals within a given
haplotype.
pre_anim_gg <- crosshap::prepare_hap_umap(umap_in,
HapObject = HapObject,
epsilon = 0.6,
vcf = vcf,
nsamples = 25)At this stage, plots for all frames for all the individuals from all
haplotypes are stacked onto a single image, so we’ll need to isolate
them. First, individuals are separated into different facets by
haplotype. Then, gganimate will stagger the images by only showing one
individual from each haplotype at a time. Finally, the animation will be
rendered with the gifski_renderer().
Feel free to experiment with the transition_length,
state_length and animate() display parameters
to find what you think best represents your data.
hap_gganim <- pre_anim_gg +
ggplot2::facet_wrap(~hap) +
gganimate::transition_states(Frame,
transition_length = 0,
state_length = 1)
gganimate::animate(
hap_gganim,
renderer = gganimate::gifski_renderer(),
fps = 3,
res = 1200,
width = 6,
height = 6,
units = "in",
#NOTE: nframes = nsamples
nframes = 25
)
gganimate::anim_save("hap_animation.gif")
What do the dots and facets indicate here?
In the GIF, the dots represent the same SNPs as in the MGumap, however now each point is coloured only if the individual possesses the alternate allele at that locus, with dark grey for SNPs unassigned to Marker Groups and light grey for reference alleles. Each facet displays individuals that share a given haplotype. The first facet shows all Marker Group SNPs for reference, it’s the same as the MGumap we built before. Individuals that are unassigned to a haplotype are placed in the final ‘Unassigned’ facet.
What you should notice is that individuals within the same haplotype will share the majority of alleles for Marker Group SNPs. However, variability is present across ‘noise’ SNPs that are not assigned to a marker group between individuals in the same haplotype group. In other words, the colours within each facet stay the same, reflecting shared haplotypes for MG SNPs, though background variation may differ.
Congratulations on building your very own haplotype UMAP GIF visualization, go show your friends!